
2026 年6月5日,实验室王红艳教授团队在心血管领域权威期刊《循环》(Circulation)发表原创研究成果,论文题为 “Aberrant Phase Separation of Endothelial MAML1 Causes Congenital Heart Disease by Suppressing Notch Activity(内皮 MAML1 异常相分离通过抑制 Notch 活性引发先天性心脏病)”。
研究团队首先在先天性心脏病临床队列中筛查到 MAML1 罕见错义突变,随后构建基因敲入小鼠、心内膜特异性敲除小鼠模型,并结合经 CRISPR 技术编辑的人类心脏类器官开展验证。研究首次证实,MAML1 可借助液 - 液相分离(LLPS)在细胞核内形成凝聚体,以此激活 Notch 信号通路;而先天性心脏病相关的电荷改变突变,或是 PKN2 激酶引发的蛋白过度磷酸化,都会扰乱这一相分离过程,阻碍心内膜 - 间充质转化(EndMT)进程,最终诱发室间隔缺损、主动脉瓣畸形等病变。该成果也正式将液 - 液相分离功能异常,认定为先天性心脏病一类全新的致病机制。
【研究背景】
室间隔缺损(VSD)是先天性心脏病里最常见的亚型,占全部病例的 30%—40%。心脏间隔与瓣膜的正常形成,高度依赖心内膜 - 间充质转化过程:心内膜内皮细胞会转化为具备迁移能力的间充质细胞,逐步填充心内膜垫,最终发育为成熟的心脏内部结构。心内膜 - 间充质转化的启动与终止受到机体精密的时空调控,一旦该过程出现缺陷,便会直接引发室间隔缺损、二叶式主动脉瓣、法洛四联症等多种先天性心脏病。
Notch 信号通路是调控心内膜 - 间充质转化的核心环节。当配体与 Notch 受体相结合后,Notch 胞内结构域(NICD)会被蛋白酶切割并释放,进入细胞核后,与 DNA 结合蛋白 RBPJ、共激活因子 MAML1 共同组装成转录复合物,进而启动HEY1、SNAI1等一系列促进心内膜 - 间充质转化的基因表达。动物实验显示,小鼠体内Maml1缺失会显著抑制 Notch 通路依赖性转录,该基因纯合缺失还会造成小鼠围产期死亡。此前已有研究发现,人类 MAML1 基因的罕见变异和二叶式主动脉瓣存在关联,但该蛋白如何通过调控 Notch 信号、影响心内膜 - 间充质转化,进而造成心脏畸形,背后的分子机制始终没有明确答案。
液 - 液相分离是细胞内一种基础的组织调控方式,蛋白质依靠弱多价相互作用形成液态生物分子凝聚体,也是转录激活过程的重要功能枢纽。现阶段已有研究证实,液 - 液相分离异常参与心力衰竭、心肌病等成人心脏疾病的发生发展,但该机制是否作用于先天性心脏病,此前尚无相关报道。
【研究方法】
本项研究从临床遗传学、动物模型、人类心脏类器官、分子机制四大维度展开系统性探索。
1.临床队列分析:选取 412 例散发性先天性心脏病患者与 213 名健康华北汉族受试者,针对发育通路关键基因开展靶向捕获测序的二次分析。
2.动物模型构建:建立对应人类 MAML1-Q401K 位点的Maml1-Q408K 基因敲入小鼠,观察小鼠心脏病理表型;利用 Npr3-CreER 系统构建心内膜特异性Maml1条件敲除小鼠,在胚胎第 8.5 天使用他莫昔芬干预,在心内膜 - 间充质转化启动前完成基因敲除;同时借助 Npr3-CreER; Rosa26-tdTomato 谱系示踪体系,直接量化心内膜 - 间充质转化效率。
3.人类心脏类器官(HHO)实验:运用 CRISPR 技术编辑人胚胎干细胞,分别构建 MAML1-Q401K 敲入细胞系与 MAML1 敲除细胞系,将其定向诱导分化为人类心脏类器官。该三维模型可自主模拟心内膜 - 间充质转化过程,研究人员据此检测类器官形态、转化相关指标以及 Notch 通路靶基因的表达水平。
4.分子机制探究:通过活细胞成像、荧光漂白恢复(FRAP)、体外重组蛋白实验,解析 MAML1 的液 - 液相分离特征;利用质谱技术鉴定 MAML1 的翻译后修饰类型及上游调控激酶;构建 S314D(磷酸化模拟型)、S314A(磷酸化缺失型)突变体,完成后续功能验证。
【关键结果】
一、MAML1 罕见错义突变与室间隔缺损存在明确关联
研究在 412 例先天性心脏病患者体内,共检出 8 种 MAML1 杂合错义突变,所有携带这类突变的患者均合并室间隔缺损,部分患者还同时患有法洛四联症。检测结果显示,这 8 种突变并不会改变 MAML1 蛋白及对应的 mRNA 表达水平,但其中 5 种突变(Q401K、T433K、E553K、N580K、M698R)可明显抑制 Notch 报告基因活性,Q401K 的抑制效果最为突出。序列比对证实,Q401 位点在物种进化过程中高度保守。
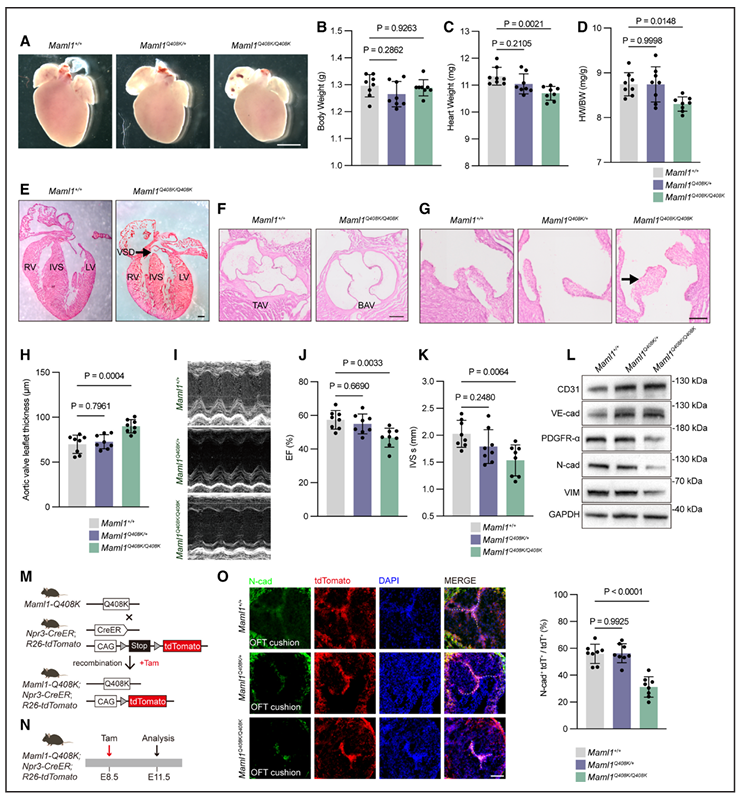
图 先天性心脏病相关的MAML1-Q408K突变会干扰瓣膜发育和EndMT,重现患者样心脏表型
二、Maml1-Q408K 敲入小鼠成功复刻人类室间隔缺损表型
对出生 0.5 天的Maml1-Q408K 纯合敲入小鼠解剖发现,其整体心脏体积偏小,心重与体重比值下降。组织学检测可见,纯合子小鼠出现室间隔缺损、二叶式主动脉瓣的概率,远高于杂合子与野生型小鼠。待小鼠生长至 6 周龄,超声心动图检测提示,其心脏射血分数降低,收缩期室间隔厚度明显变薄。
在胚胎第 11.5 天的心脏内皮细胞中,内皮标志物 CD31、VE-cadherin 表达上调,而 PDGFR-α、N-cadherin、波形蛋白等间充质标志物表达下降,Notch 通路靶基因Hey1、Snai1的表达也同步下调。谱系示踪数据进一步显示,相较于野生型小鼠,Q408K 纯合子流出道心内膜垫区域,发生心内膜 - 间充质转化的细胞占比显著降低。
三、心内膜特异性敲除模型、心脏类器官验证内皮细胞的致病作用
出生 0.5 天的心内膜特异性Maml1敲除小鼠,同样出现心重 / 体重比下降,室间隔缺损、二叶式主动脉瓣发生率升高等问题。6 周龄小鼠的心脏功能、室间隔形态改变,以及各类标志物的表达变化,均与Maml1-Q408K 敲入小鼠表现一致。
从 MAML1 敲除人胚胎干细胞分化而来的心脏类器官,培养至第 10 天时体积明显小于野生型。细胞表型上,内皮标志物表达升高、间充质标志物表达受抑,HEY1、SNAI1基因表达显著下调。而引入 MAML1-Q401K 突变的心脏类器官,也呈现出完全一致的表型:类器官发育受阻、心内膜 - 间充质转化过程被抑制、Notch 靶基因表达下降。动物模型与类器官的实验结果相互印证,明确 Q401K 为致病性突变。
四、MAML1 依靠 IDR2 结构域实现液 - 液相分离
经 GFP 标记的 MAML1 蛋白,可在血管内皮细胞的细胞核内形成动态点状凝聚体,凝聚体之间还能发生自主融合。荧光漂白恢复实验显示,荧光信号可在 30 秒内恢复约 65%,符合液态凝聚物的典型特征;使用 1,6 - 己二醇处理后,细胞核内的凝聚体彻底消失。体外实验中,经纯化的 MAML1-GFP 蛋白在 PEG 诱导下可形成球形液滴,具备融合能力,荧光恢复速度快,同时对高盐环境敏感,证明 MAML1 的液 - 液相分离过程依赖静电相互作用。
内在无序区(IDR)是 MAML1 蛋白的主要组成部分。研究人员将该区域划分为四个片段后发现,单独缺失 IDR2 片段,会大幅削弱蛋白形成液滴的能力,其余片段缺失则几乎不会造成影响。与之相对,仅保留 NICD 结合域与 IDR2 的精简结构,就足以形成液滴,且同样具备快速荧光恢复、高盐敏感的特性,由此确定IDR2 是调控 MAML1 液 - 液相分离的核心元件。
功能实验表明,利用 1,6 - 己二醇破坏 MAML1 的相分离后,蛋白与 NICD1 的结合能力减弱,HEY1、SNAI1等 Notch 靶基因的表达随之降低。缺失 IDR2 的突变体基本丧失了激活 Notch 信号的能力;而仅保留 NICD 结合域与 IDR2 的精简片段,可部分恢复 Notch 通路活性。这说明,MAML1 必须借助 IDR2 介导的液 - 液相分离,才能高效结合 NICD1 并启动基因转录。
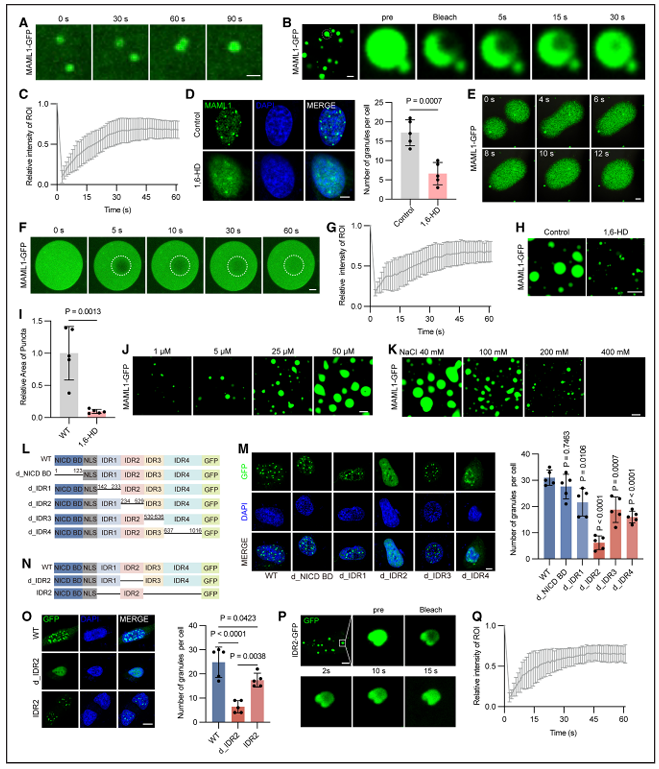
图 MAML1的相分离由内在无序区域介导,调控Notch信号传导
五、先天性心脏病相关突变破坏 MAML1 相分离与 Notch 通路活性
此前筛选出的 5 种可抑制 Notch 活性的突变里,Q401K、T433K 两个位点均位于 IDR2 区域。对Maml1-Q408K 纯合小鼠心脏内皮细胞观察发现,细胞内 MAML1 凝聚体数量明显减少。体外蛋白实验进一步验证,携带 Q401K 突变的 MAML1 蛋白,形成的液滴数量少、流动性差,荧光漂白恢复速率慢于野生型,蛋白与 NICD1 的结合效率也大幅下降。
六、PKN2 介导 MAML1 S314 位点磷酸化,负向调控液 - 液相分离
质谱分析在 MAML1 的 IDR2 区域鉴定出 S314、S360 两个磷酸化位点,其中 S314 是主要修饰位点。构建磷酸化模拟突变体 S314D 后发现,该突变会抑制 MAML1 相分离,降低蛋白与 NICD1 的共定位及结合水平,Notch 通路活性随之下降;而磷酸化缺失突变体 S314A,则能增强相分离效果,提升 Notch 信号活性。
研究通过质谱筛选 MAML1 免疫共沉淀复合物中的核定位激酶,PKN2 的匹配度最高。使用 PKN2 抑制剂 PKN1/2-IN-1 处理细胞后,MAML1 S314 位点的磷酸化水平显著降低。细胞过表达 PKN2,会加剧 S314 磷酸化、抑制蛋白相分离;利用 siRNA 敲低 PKN2,则会减少磷酸化修饰、促进凝聚体形成。体外重组蛋白实验最终证实,PKN2 可直接对 MAML1 S314 位点进行磷酸化,并通过这一方式抑制液 - 液相分离。
结合心脏发育的时间规律来看,在心内膜 - 间充质转化的关键窗口期(胚胎第 11.5 天),流出道心内膜垫内皮细胞内 PKN2 表达水平偏低,MAML1 S314 磷酸化程度弱,以此维持 MAML1 凝聚体稳定存在,保障 Notch 信号正常传导。随着胚胎不断发育,PKN2 表达逐步升高,S314 磷酸化随之增强,MAML1 凝聚体逐步解离,Notch 靶基因表达下调,最终实现心内膜 - 间充质转化过程的适时终止。
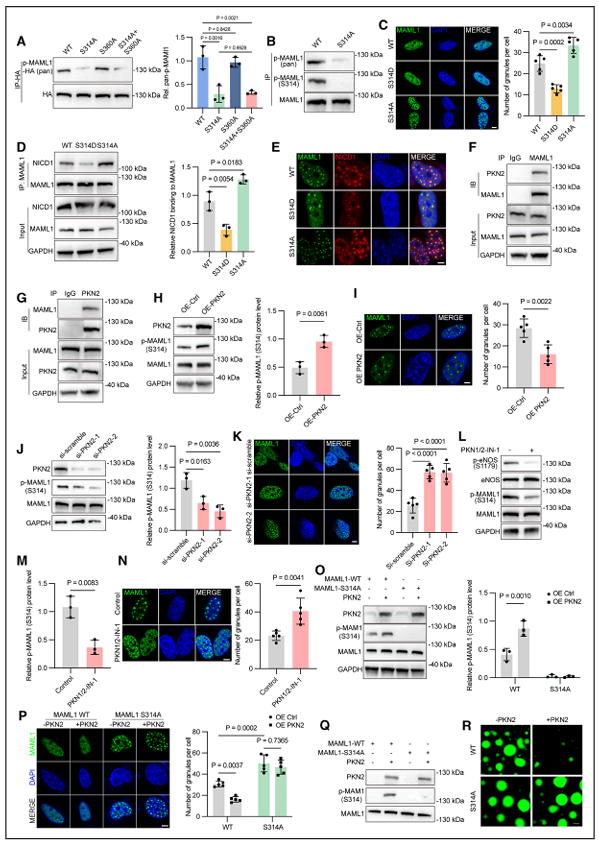
图PKN2介导的MAML1在S314处的磷酸化调控其相分离和缺口信号活性
七、PKN2-MAML1 S314 信号轴调控人类心脏类器官发育
由携带 MAML1-S314D 突变的人胚胎干细胞分化而来的心脏类器官,体积小于野生型,心内膜 - 间充质转化过程受抑,Notch 靶基因表达下调;而 S314A 突变型类器官体积更大,转化过程明显增强。
在野生型类器官中过表达 PKN2,会阻碍类器官正常发育,但该操作对 S314A、S314D 突变型类器官无明显作用,证明 PKN2 发挥功能完全依赖 MAML1 的 S314 位点。
【结论】
MAML1 蛋白 IDR2 区域的静电状态,是心脏发育过程中整合遗传突变、翻译后修饰两类信号的关键节点。MAML1 依托 IDR2 介导的液 - 液相分离在细胞核内形成凝聚体,这也是蛋白高效结合 NICD1、激活 Notch 转录程序的结构基础。先天性心脏病相关的电荷改变突变(Q401K、T433K),以及 PKN2 激酶诱导的 S314 位点过度磷酸化,都会破坏 MAML1 正常的液 - 液相分离,减弱 Notch 信号传导,造成心内膜 - 间充质转化受阻,进而引发室间隔、主动脉瓣畸形。该研究明确,液 - 液相分离功能异常是先天性心脏病一类全新的致病机制。
【临床意义】
研究证实,MAML1 的 IDR2 区域是先天性心脏病致病突变的高发区域,而酸性氨基酸被碱性氨基酸替换引发的电荷改变,是这类突变致病的核心特征。在先天性心脏病产前筛查与产后遗传诊断工作中,可优先对该区域的错义突变进行检测,有助于提升室间隔缺损、二叶式主动脉瓣的分子诊断准确率。
PKN2 作为负向调控 MAML1 相分离的激酶,其活性直接决定心内膜 - 间充质转化窗口期的开启与关闭。细胞及类器官实验均证明,PKN2 抑制剂可恢复 MAML1 凝聚体形成能力,重新激活 Notch 信号。虽然距离临床落地还有较长距离,但 PKN2-MAML1 信号轴的发现提示,调控生物分子凝聚体稳定性,而非单纯改变 MAML1 蛋白表达量,有望成为先天性心脏病全新的干预思路。
此外,本研究所用的人类心脏类器官模型,可自主模拟心内膜 - 间充质转化过程,无需额外外源诱导,规避了传统体外模型的实验偏差。该平台也可用于先天性心脏病致病基因的快速功能验证,以及候选治疗药物的初步筛选。
【研究局限】
第一,本次在先天性心脏病患者队列中,MAML1 突变整体检出率偏低,412 例样本中仅检出 8 例(约 1.9%),仍需更大规模、多中心的临床队列,进一步验证该基因突变在疾病发生中的作用。
第二,除 Q401K 外,T433K、E553K、N580K、M698R 这几类突变尚未在动物模型中逐一验证,它们在活体动物体内的致病性还需后续实验补充证实。
第三,全身敲除 PKN2 会导致小鼠胚胎在第 9.5—10 天死亡,因此还需要借助条件性敲除模型,深入解析 PKN2-MAML1 轴在心内膜 - 间充质转化阶段的时序特异性调控作用。
第四,目前关于液 - 液相分离的机制探索,大多基于体外实验与细胞水平研究,MAML1 凝聚体在活体胚胎心脏内的动态变化,仍缺少直观的观测证据。
原文链接:https://www.ahajournals.org/doi/full/10.1161/CIRCULATIONAHA.126.078188
【文章来源:365医学网】

版权所有:复旦大学复杂性状的遗传调控全国重点实验室
Copyright: State Key Laboratory of Genetics and Development of Complex Phenotypes